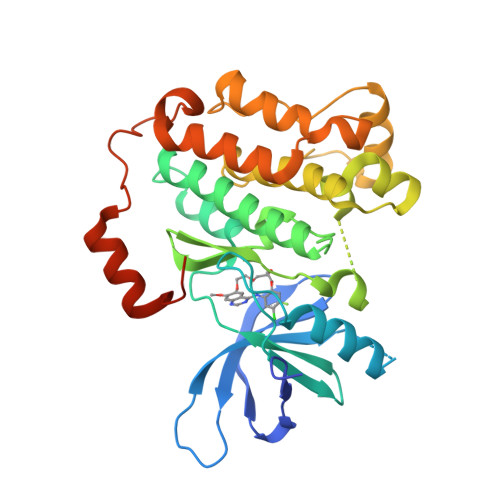
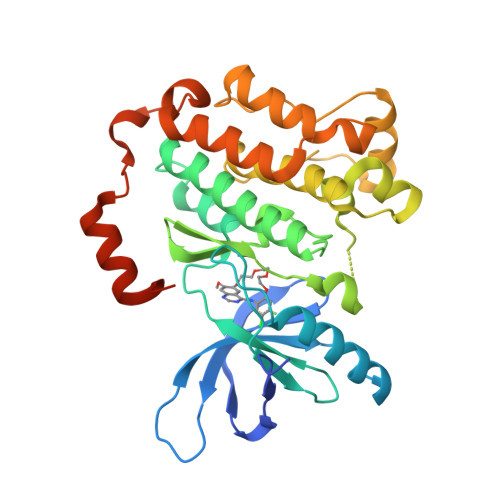
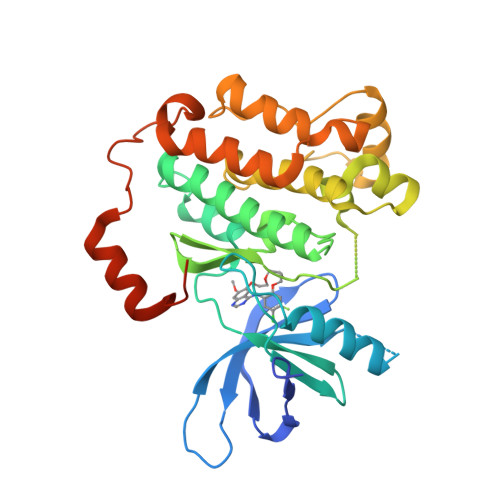
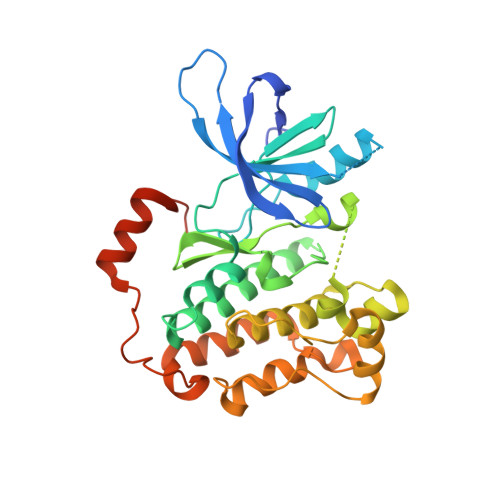
Macrocyclization of Quinazoline-Based EGFR Inhibitors Leads to Exclusive Mutant Selectivity for EGFR L858R and Del19.
Amrhein, J.A., Beyett, T.S., Feng, W.W., Kramer, A., Weckesser, J., Schaeffner, I.K., Rana, J.K., Janne, P.A., Eck, M.J., Knapp, S., Hanke, T.(2022) J Med Chem 65: 15679-15697
- PubMed: 36384036
- DOI: https://doi.org/10.1021/acs.jmedchem.2c01041
- Primary Citation of Related Structures:
7U98, 7U99, 7U9A - PubMed Abstract:
Activating mutations in the epidermal growth factor receptor (EGFR) are frequent oncogenic drivers of non-small-cell lung cancer (NSCLC). The most frequent alterations in EGFR are short in-frame deletions in exon 19 (Del19) and the missense mutation L858R, which both lead to increased activity and sensitization of NSCLC to EGFR inhibition. The first approved EGFR inhibitors used for first-line treatment of NSCLC, gefitinib and erlotinib, are quinazoline-based. However, both inhibitors have several known off-targets, and they also potently inhibit wild-type (WT) EGFR, resulting in side effects. Here, we applied a macrocyclic strategy on a quinazoline-based scaffold as a proof-of-concept study with the goal of increasing kinome-wide selectivity of this privileged inhibitor scaffold. Kinome-wide screens and SAR studies yielded 3f , a potent inhibitor for the most common EGFR mutation (EGFR Del19: 119 nM) with selectivity against the WT receptor (EGFR: >10 μM) and the kinome.
Organizational Affiliation:
Institute of Pharmaceutical Chemistry, Goethe University Frankfurt, Max-von-Laue-Straße 9, 60438 Frankfurt, Germany.