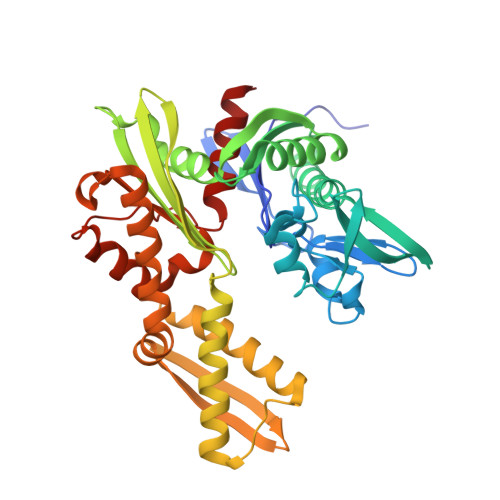
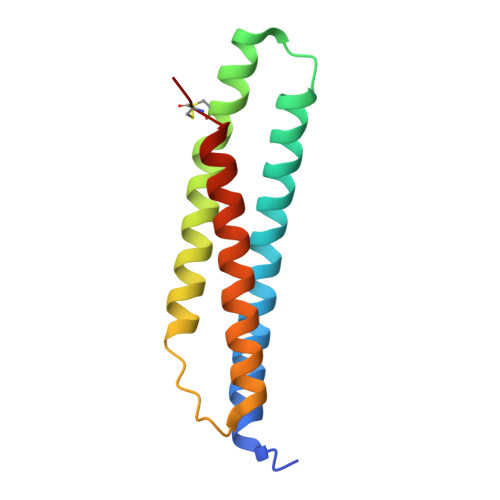
A fragment-based approach applied to a highly flexible target: Insights and challenges towards the inhibition of HSP70 isoforms.
Jones, A.M., Westwood, I.M., Osborne, J.D., Matthews, T.P., Cheeseman, M.D., Rowlands, M.G., Jeganathan, F., Burke, R., Lee, D., Kadi, N., Liu, M., Richards, M., McAndrew, C., Yahya, N., Dobson, S.E., Jones, K., Workman, P., Collins, I., van Montfort, R.L.(2016) Sci Rep 6: 34701-34701
- PubMed: 27708405
- DOI: https://doi.org/10.1038/srep34701
- Primary Citation of Related Structures:
5AQF, 5AQG, 5AQH, 5AQI, 5AQJ, 5AQK, 5AQL, 5AQM, 5AQN, 5AQO, 5AQP, 5AQQ, 5AQR, 5AQS, 5AQT, 5AQU, 5AQV, 5AQW, 5AQX, 5AQY - PubMed Abstract:
The heat shock protein 70s (HSP70s) are molecular chaperones implicated in many cancers and of significant interest as targets for novel cancer therapies. Several HSP70 inhibitors have been reported, but because the majority have poor physicochemical properties and for many the exact mode of action is poorly understood, more detailed mechanistic and structural insight into ligand-binding to HSP70s is urgently needed. Here we describe the first comprehensive fragment-based inhibitor exploration of an HSP70 enzyme, which yielded an amino-quinazoline fragment that was elaborated to a novel ATP binding site ligand with different physicochemical properties to known adenosine-based HSP70 inhibitors. Crystal structures of amino-quinazoline ligands bound to the different conformational states of the HSP70 nucleotide binding domain highlighted the challenges of a fragment-based approach when applied to this particular flexible enzyme class with an ATP-binding site that changes shape and size during its catalytic cycle. In these studies we showed that Ser275 is a key residue in the selective binding of ATP. Additionally, the structural data revealed a potential functional role for the ATP ribose moiety in priming the protein for the formation of the ATP-bound pre-hydrolysis complex by influencing the conformation of one of the phosphate binding loops.
Organizational Affiliation:
Cancer Research UK Cancer Therapeutics Unit, Division of Cancer Therapeutics, The Institute of Cancer Research, London SM2 5NG, United Kingdom.